Back from a friend’s party where you tried homemade beer? And now you want to try yourself right? I see. Well, here we will (only) go through the details of getting the so-called “pitching rate” (or initial cell density) using a hemocytometer. Try not to be drunk when you do this. I am serious – you’re using glass material after all! For more info on the basic materials you will need to count yeast cells, check our cell counting starter kit post and our beer making supplies post.
1. Taking a representative sample of yeast
If you want to count cells it’s certainly because you already started a culture. So go get 1mL for your count. Make sure you mix properly before taking the sample, it should be as homogeneous as possible.
2. Adding viability dyes
If you’re interested in the healthiness of your yeast, you should consider adding a viability dye. A viability dye (such as methylene blue) will penetrate cells that are dead but will be excluded (that’s the PhD word) from live ones – so you can count both populations separately and know the percent of live over total cells.

It is common to dilute directly in a methylene blue solution (0.1%) but you can also add one drop of the original solution as long as the volume of cells you have is quantitatively bigger (for example, it won’t make a difference in terms of dilution to add 1 drop to 10mL – i.e. you can do it). See step 3 for the dilution part.
You should wait a few minutes for the dye to react. Methylene blue is oxidized in live cells but remains unchanged in dead cells. Be aware that viabilities under 90% might not be accurate due to the metabolic origin of the reaction. Check the viabiliy dyes post for alternatives.
3. Diluting the sample
This step is quite subjective. You will get better with time, but in the beginning it’s just trial and error.
The idea behind this is that you don’t want to have too many cells in the small space where you are going to count (they would overlap and you would get a headache trying to figure out which is which), but you need to have enough (at least 100) for your count to be significant. Let’s say you decide to dilute your 1mL in 9mL (if you haven’t done it yet with your methylene blue solution). You should do steps 4&5 to check that there are at least 100 cells in the central big square, but they are countable. If you decided to go for the methylene blue dilution in step 2, then that’s your diluting solution. Otherwise, do it with distilled water.
4. Preparing the hemocytometer and adding the sample
To prepare your hemocytometer, make sure that the chambers and the coverslip are clean (use water or ethanol, and a soft tissue). Place the coverslip on the hemocytometer, on top of the two coverslip supports on the sides of the overfill chambers.Mix your sample well and pipette a small amount into both chambers, avoiding to push the fluid too hard; it should go in almost on its own by capillarity.
5. Counting the yeast in the central square of the hemocytometer
Yeast cells have an average size of 5-10μm. In comparison to the size of a hemocytometer square (1mm), they are 100-200 times smaller. So you should count the smaller squares in this case (i.e., the ones in the central square, which measure 25 x0.2mm). They also have helper lines that divide each of them into 16 even smaller squares.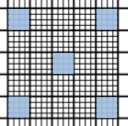
Count 5 of the 0.2mm squares, evenly distributed throughout the central square (commonly, the four corners and the central one). The rules are the same as for the other uses of the hemocytometer: establish beforehand two sides (lines) that you are not going to count (bottom and right for example) and keep those consistently for all the 0.2mm squares. Count the live and the dead cells and record your counts somewhere (or use this awesome app for iPhone/Android).
An important remark however is that yeast duplicates by budding (creating a bump that becomes bigger and finally results in a second, independent cell). It is common practice to count cells that haven’t divided yet (even if they have the bump) as one, not two. This is because there are different levels of “buddyness”, so your choice for counting one or two would be biased.
6. Practice the counting
There’s this great site that generates hemocytometer squares for you – so you can practice the counting before getting hands on!
7. Performing the calculations
Perform the calculations as in hemocytometer calculation, except that you have to multiply by 10000*25 = 250000 (because there are 25 small squares in a 1mm wide square) to get the cell density. There’s a couple of tools that will make your life SO MUCH easier when doing hemocytometer calculations:

|

|
Thanks for taking the time to post! I found this very helpful.
Thank you so much for this post. It’s very helpful for my yeast populating growth lab! I sure will cite this 🙂 haha
Glad to be of help!! Let me know of any questions you might have.
Hi Since I am never a math person, I am kind of stuck with the calculations part. If I am counting only five squares (1mm area with 16 0.2mm smaller squares), how should I do my calculations if I am only taking the sample? Like if I want the number for the whole central square? Thank you (in advanced) 🙂 !
Hi Joanne,
Ok first thing, you need to take the average cell count in the small squares, meaning that the base unit you will be using will be a small square (and therefore you need the volume of a small square only). It does not matter that you’re not counting all of your cells (that’s the beauty of a representative sample); your final result is going to be a cell density, therefore it represents a concentration as x number cells in 1 smaller square or 25x cells in 25 smaller squares (it’s exactly the same thing).
There are two ways the volume of a small square can be calculated: either from the volume of a big square or from its own measurements.
So, if you’re counting the 1mm central square with 0.2mm small squares, there are 25 of those squares (not 16). In this case, the volume you have to divide by is 0.000004 mL, because the surface of one of these squares is 0.2mm x 0.2mm = 0.04mm2 (see here for a detailed conversion to volume). You can also deduce it from the volume of the central square (which is 0.0001mL) as: 0.0001mL/25 = 0.000004mL.
If you’re counting a 1mm corner square with 0.25mm small squares, there are 16 of those. In this case, the volume you have to divide by is 0.00000625 mL, because the surface of each of them is 0.25mm x 0.25mm = 0.0625mm (see here for a detailed conversion to volume). You can also deduce it from the volume of the corner square (which is 0.0001mL) as: 0.0001mL/16 = 0.00000625mL.
Finally, you should go to hemocytometer calculation and perform your calculations with the volume of a small square calculated as above.
Hope that helped!!!
Thank you so much!!! You’re amazing, I finally understand the whole thing :D. My bio teacher is going to be proud of me! Thank you, again!! :))
Great that we made it! Come back if you have more questions anytime. Good luck with your bio 🙂
Hi Just a question! I incubated hanseniaspora in 10ml broth->centrifuged them -> add hanseniaspora pelletes into 9mL peptone+ mixed -> 1mL of mixture and 2mL of methylene blue were mixed. then I counted 5 squares, and I got 102, 120, 128, 145, 133. From here how can I calculate the total numbers of hanseniaspora. could you please help me? 🙁
I’m not sure whether I should think dilution factor to be 1:3 (just counting the methylene blue) or whether to include dilution of peptone + methylene blue dilution
Hi Pauline,
Ok let’s do it step by step. First of all let’s define the solutions you’re preparing:
A: 10mL of broth (original solution)
B: 9mL of peptone containing all the yeast (but no liquid) from A
C: 1mL of B + 2mL of methylene blue (3mL of counting solution)
You’re performing the count on solution C so let’s do the calculations backwards. From your hemocytometer counts, you get an average of 125.6 cells in each small square. The cell density in C is calculated as: 125.6 x 250,000 = 31.4 million cells/mL. Because you have 3mL of this solution, the total number of cells in C is 31.4 x 3 = 94.2 million cells.
The density of solution B will be the same as the one in the 1mL you have taken to count. Since adding methylene blue doesn’t change the number of cells, just the density, you will have the same number of cells in that 1mL than in the 3mL of solution C (94.2 million cells). The density in that 1mL is 94.2 million cells/1mL = 94.2 million cells/mL. That is the density of B, too. But there’s 9mL in total of solution B, so the total number will be 94.2 million cells/mL x 9mL = 847.8 million cells. If you only want to know the ones that are left after removing the 1mL (in the remaining 8mL): 94.2 million cells/mL x 8 mL = 753.6 million cells.
Finally, in order to know what was the density in the original solution A, observing that the number of cells is the same as in B (before removal of the 1mL), the density in A will be 847.8 million cells / 10mL = 84.78 million cells/mL.
As a rule of thumb, remember that adding volume of a liquid that doesn’t contain yeast to a solution that contains yeast will only change its density (the total number will stay the same). However, removing volume from a solution that contains yeast will decrease the total cell number (but the density will stay the same if it is properly mixed).
Hope that was helpful! Let me know of any other questions you might have 🙂
Maria
Hello, this was very helpful, however, I do not know how to calculate the percentage recovery after washing my yeast cell off the haemocytometer, that’s the post-washing. I am being asked to Calculate the percentage recovery. Thank you.
Hi Mfonabasi,
I am not familiar with any technique that involves washing yeast off the hemocytometer, and then calculating how many cells you recover from there. Could you please provide the full question that was asked so that I can try to understand a bit better the context? Thanks!
And sorry for not being of help straight away.
Maria
A hearty thumbs up for a great explanation of the process–very much appreciated, from a biology teacher always looking to make things a little clearer. I’ll be using and referencing your site for this protocol. Thanks.
Thanks so much! I’m always happy to have positive feedback 🙂 But do let me know if you have any ideas on things I could improve. Happy teaching!
thank you for the website. great information!! Which microscope objective (zoom) is best recommended to count yeast cells?
Hi Sergio, I’m glad it was helpful 🙂
You’d need a 400X magnification to see the small squares within the central square (for the counts) and 100X to see all squares within a hemocytometer chamber (for an overall view). I’d say this microscope is a good candidate, although you can always shop around and look for your favorite features (binocular models are a lot more comfortable, a mechanical stage helps move the hemocytometer when counting, etc…) which can sometimes be added later on.
Btw, if you have an LCD microscope, I recommend ImageJ to archive the images. There is also a cell counter add-in that lets you mark and count the cells on the pictures. Actually, there are a slew of useful scientific add-ins. It’s a great for archiving. I have no affiliation with this free software, I just use it for saving real images to help teach counting technique.
That’s a great suggestion, thanks Ken! I believe the plugin you’re referring to is this one and ImageJ can be downloaded from here. I’ve used ImageJ in the past for measuring particles in TEM images but never for cell counting. I believe this can be a really easy way to save time when counting cells if you have a microscope connected to a computer.
Thanks again!
Maria
Thank you for posting this! I’m studying A level in Biology and i found this very useful! 🙂
Cool!! Good luck with your exams 🙂
How can you determine what dilution is the most accurate to calculate cell concentrations for your original sample, after you have calculated the Cell density???
Hi Samuel,
Good question! So once you have a cell density from the first count (let’s call it CD), you go back to the basics: the minimum number of cells to count is 100. If you’re doing the count for yeast cells, you’ll be doing it on the central square so you’ll want to have a cell concentration of: 100/5 x 250,000 x dilution factor = 5,000,000 x dilution factor. Now, you equal the cell density from the first count to the equation and you get: CD = 5,000,000 x dilution factor. Or, dilution factor = CD/5,000,000. Let’s say your CD is 100,000,000 cells/mL, then you will need to dilute 20 times, in other words, add 10uL of original sample to 190uL of diluting solution/methylene blue/erythrosine (or do serial dilutions). Bear in mind that this is approximative – if you want to be confident you’ll get slightly more than 100 cells in the central square, then your dilution has to be 20 (in the example) or less.
I hope that answers your question – otherwise ask again!
Cheers
María
Thank you I’m a bit confused maybe I should give you my senerio. I have conducted a Serial cell dillution with 100ul of cell suspension but only added 50ul then diluted it with 50ul of cell medium and then added 50ul of the Trypan overall each dilution has a volume of 100ul, with the dilution factor starting at 1in4,1in8, 1in16, 1in32 and 1in64 I have calculated the density of the total cells but from the result I have got I want to know how to find out which cell density I have got is the most accurate dilution. Thank you, hope you can help because I’m really struggling to get my head around it
OK, I understand now. You have the cell density values for all dilutions. And you want to know which of them is the most accurate. Let’s take the question the other way round: which cell densities are not so accurate? That’s called an outlier. What you should do is enter your data here and then the ones that come outside the whiskers are considered outliers (it will even say it’s an outlier: try with this set 100000,125000,120000,110000,90000,250000). Anything inside is considered to be accurate values, there’s no one single answer, but a range (measured by the confidence level).
Hope that helped! Cheers
Maria
Thanks you so mcuh, that’s very very helpful
Not a problem! Happy counting 🙂
NICE to read it ,,, found it helpfull
sir please confirm why we should multiply 25 with 10000
Hi there,
10,000 is the inverse of the volume of the whole central square (in mL-1, see here for details on where the number comes from). 25 is the number of small squares inside the central square. If you have counted say 5 small squares and have taken the average, then you have the average number of cells in one of those 25 so for the whole central area you have to multiply by 25. Hope that helps!
Cheers
Maria
sir thank you for the reply…………. bt still iam confused bcz as you mentioned there volume of one small cell is comes to 0.000004mL if we convert it for 1mm it wont come 10000 please brief this sir
thank you
Sure. So 0.000004mL is the volume of one small square inside the central square. You can use that number as is to calculate the density, i.e. if you use the formula:
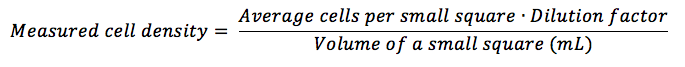
You can put directly the 0.000004mL in the bottom section of the equation. A different approach is: 1/0.000004mL = 250,000 right? Similarly, 1/0.0001mL = 10,000 so in order to calculate the density of cells you can either multiply the average number of cells per small square by 250,000 or by 10,000 x 25 (because there are 25 small squares in the central square, so the 25 is to convert the average number of cells in one small square into the average number of cells in the central square) which is exactly the same. Was that clearer?
Hi,
I need to prepare 40ml Candida suspension at final density of 1000000 cells/ml. I inoculated RPMI medium with loopful of cells, harvested from SDA plate. How much of this suspension to pippete into both chambers? Should I count in the central square into both chambers?
I count 5 of the 0.2mm squares, evenly distributed throughout the central square (commonly, the four corners and the central one). What is the formula of cell density and how to prepare 40 mlCandida suspension at final density of 1000000 cells/ml?
Thank you in advance!
Milica
Hi Milica,
You should pipette around 10uL of fluid into each chamber, but make sure you do not overfill the chamber. Once the liquid reached the limits of the coverslip, stop filling the chamber. The fluid to use depends on what kind of count you want to perform. If you just want cell number, you can use cell suspension straight from the harvested ones (you might need to dilute). If you want viability too, then you should use viability dies at 1:1 to 1:5 ratio to cells. The ideal is to have a counting suspension that allows you to count around 100 cells per chamber, in your case this would be in the 5 small squares you will count.
The formula for cell density is (more info on hemocytometer calcs here):
cell density = average cells counted in 5 small squares x dilution factor x 250000
with:
average cells counted in 5 small squares = total cells counted in all 5 small squares / 5
Once you have the current cell density (which is given by the formula above based on hemocytometer counts), you have to find out how much volume you need to take from your old suspension:
current cell density x volume to take = final cell density (1,000,000) x final volume (40)
or:
volume to take = 1,000,000 x 40 / current cell density
Once you get your current cell density as measured by hemocytometer, you can calculate the volume to take with the formula above. To prepare the final solution, mix that volume of original cell suspension with medium (the difference with the volume of cell suspension taken, up to 40mL) and that’s it.
Also, you should count the central square in both chambers for improved accuracy.
Hope that helps!
Maria
Dear Maria,
Thank you very much for your reply and excellent explanation. I need to count Candida cells in RPMI and to use that suspension for biofllm formation. I inoculate RPMI with Candida harvested from SDA plate, vortex it and count. What do you think – is it necessary to dilute it before counting?
Thanx in advance!
Milica
Dear Maria,
I have one more question.
Is the formula: cell density = average cells counted in 5 small squares x dilution factor x 250000 valid only in case if I count the cells in 1ml of medium (if 1ml of media is inoculated with fungi harvested from agar)? Generally, it is not enough amount to prepare 40ml of medium with concentration of 1,000,000 cell/ml. If I suspend cells in 5ml of medium (without dilution) should I use the same formula?
I am still confused with this.
Thank you very much in advance,
Milica
Hello,
Could you tell me please what is the formula for average cell density if I have 5ml suspension of fungi (5ml of medium was inoculated with loopful of cells harvested from SDA agar). From this 5ml I need to prepare 20ml of medium with concentration of 1,000,000 cell/ml. Could you help me?
Thank you very much in advance!
Milica
Hi Maria,
Great website with a lots of useful information. Do you know if Trypan blue can be used to stain yeast cells? I know methylene blue is most commonly used but has some major drawbacks. I have a science background as well and have tried looking up papers with a protocol without any luck.
Thanks!
Hi Chris,
Good to hear you find the blog useful!
Yes it can, I actually found a report from Beckman Coulter comparing the efficacy of trypan blue vs methylene blue in Saccharomyces cerevisiae. I hope that satisfies your scientific rigor! 🙂
Let me know if you have any other questions.
Maria
PS: here’s the link 😉
https://www.beckmancoulter.com/wsrportal/bibliography?docname=Ta-204.doc
Thanks for the link Maria, I did actually see that report but was a bit disappointed with the fact that there was no methods section describing how they used the dye. I read a paper today that said resuspending the yeast in PBS pH 7.4 would help the dye stain the cell wall so I tried this and it worked well. In my previous attempts at using Trypan Blue with yeast I resuspended in water.
I write the “Science and Homebrews” blog for the homebrewing website Byo.com. Any chance you would like to work on putting together a blog post with me to get the readers of my blog access to your content? If so, send me an email at sciencehomebrews@gmail.com.
Thanks again!
Hi Chris,
It’s great you managed to find an article with methods that works for you! I actually did see this one but I don’t have access (I’m not in academia anymore!). Maybe you can have a look and see if they report something else, in case you need it.
I will send you a separate email on your proposal to write a blog post together.
Thanks!
Maria
Hi!
I study biology and at my school we are doing a project about yeast. I just cant find what the desireable viability is for yeast. I mean what is good yeast? How do you know if your yeast is viable, when is it viable? If 60% of the yeast is alive of if 90% of the yeast is alive?
Hope you can help me!
Hi Anki!
Viability represents the percentage of cells that are alive over all cells. I used to culture human cells and great viabilities were around 95%; 90% was good; 80-90% was decent and anything below 60% was poor. I think it is pretty similar for yeast. That doesn’t mean that it won’t grow again, it just means that something is wrong with the culture or the processing of the cells.
To assess viability, you can use the hemocytometer with viability dyes. See my review of viability dyes and my hemocytometer protocol for details of how viability is measured and calculated.
Let me know if that clarifies 🙂
Maria
Hi Maria,
Thank you! That helps alot 🙂
Hola Maria. This is an awesome website, thank you!! The app works great, I bought your Cell culture for newbies book and its great too! This is a good article. Can you tell me where I can find the steps that are being referenced. For example towards the end under: Diluting the sample it says “If you decided to go for the methylene blue dilution in step 2”. I may be missing it but where is step 2, is that in another article? Muchisimas Gracias
Hola Plutarco,
Really glad you find the app, the blog and the book useful. I have added the step numbers to the article titles, they went missing for some reason. I hope that clarifies, otherwise please let me know.
Saludos,
Maria
Hi,
When using a camera with a microscope, what camera resolution do you recommend to be able to count yeast cells? And also to be able to use the image for automatic cell count?
Regards,
Vasco.
Hi Vasco,
It depends on the microscope resolution. Here‘s an article explaining the pairing between camera resolution and microscope magnification. You typically count yeast cells under 400X so you’ll need a higher resolution than what comes in that table. Looking on Amazon, it looks like the cameras that come with 400x microscopes are 1.3MP-5MP (see this one for example).
You should be able to use the camera for cell count, try this plugin. I’d also take a look at Oculyze to forget all the hassle of buying a camera, setting it up and analyzing images.
Let me know if you have other questions.
Cheers,
Maria
Hi Maria, thank you for your reply.
I’ve seen Oculyse and we got a quote for it, and unfortunately it’s quite expensive. For the same cost we mount a whole lab 🙂
We are setting up a small lab at the brewery in order to quickly check yeast viability before pitching from fermenter to fermenter. Also to see if there’s any contamination. Simple things which we can do for considerable less cost than Oculyze, and still fairly automated. We also get added flexibility and with the difference in price we can get other tools. Your suggestion seems to be the way to go for us.
Regards,
Vasco.
Hi Vasco,
This is Kilian from Oculyze. Thank you for the feedback, we always appreciate hearing from others about our product! Sorry to hear you found our automatic microscope too expensive. Have you thought about the time it is going to take you to setup the microscope with the plugin? Once you have your sample prepared, a cell count and viability analysis with Oculyze takes one minute, with documentation included. Doing the same analysis and documentation with a microscope will take you at least 10 minutes and you will have a great potential for human error and inter-operator variability.
Our device has been validated and tested (http://www.oculyze.de/files/validation_oculyze_vlb.pdf) by the VLB (research institute for breweries in Germany) against automated devices costing many times as much . The device is guaranteed to work, this is why we offer a 30 day free trial and if you are unsatisfied, you ship it back and don’t pay for the device.
Kilian
PS;:forgot to mention- the microscope in the amazon link uses a 1.3 mega pixel camera ours has 8 mega pixels.
Hi Kilian. I was a bit surprised for finding this message. I didn’t think this was the right arena for such a commercial discussion, and now I feel obliged to clarify the context.
When I said setting up whole lab, I was talking about a simple small scale lab to be able to check basic parameters that all brewers should be checking. My apologies if it sounded different. It was just emphasizing how surprised I was regarding the cost and what can be done for the same or less money. Even if with more hassle.
I first saw Oculyze on the IBD magazine early this year and thought it was cool. Therefore the interest, and tried to find more about it. Not only it is neat, small and portable, but we are also tech people from engineering and IT and of course are drawn to clever gadgets. However, I was quite surprised by the cost, and one will always be dependent on your services as well.
When looking for alternatives, and because there are a few options, I ended up with a few questions in order to organize ideas. I had already found a few Microscopes, but needed to narrow down the options and be really sure to make an adequate choice. This arena proved useful and I have to thank Maria once again.
This one https://www.amazon.com/dp/B01BPJJL0Y?psc=1#feature-bullets-btf with a 5MP camera.
Alternatively, with the current mobiles we can also easily shoot a photo through the ocular. Meaning that even a simpler and cheaper microscope is suitable.
With a microscope one also gets added flexibility using it for other tasks. It’s also a challenge, an opportunity to develop a new set of skills. And implementing a script is also part of that challenge. Although certified in brewing, it shouldn’t come as a surprise that with a home brewing background one is used to challenges. And 10 minutes is hardly an eternity. And there’s always a few extra pair of hands around.
Don’t get me wrong, Oculyze seems impressive impressive and I do wish you the best. But it belongs in different context. As I said, the same or less cost allows a small modest brewery to buy considerable more equipment for more tasks than just checking yeast viability, even if with a bit more hassle and it takes slightly longer. And one learns in the process.
Best Regards,
Vasco.
Hey Vasco,
Thanks for taking the time to write such a detailed reply!
I just wanted to highlight the differences, also for other readers who might see your post.
Our device might be a bit more expensive then a low end microscope, but it is a lot cheaper than other automated systems, while giving more accurate results and managing your data.
If you don’t want a fully automated solution and prefer to tinker and set up your own in house system, we totally understand. As you can imagine, we as a team of developers and entrepreneurs are also personally very much tinkerers ourselves.
Good luck with your brewery!
Cheers,
Kilian
I wanted to know I am counting cells of drosophila larvae hemolymph. I am counting four corner squares , average cell count is 28 what will be the total amount of cell in 80 microlitre if I am mixing the 5micro litre sample with 5 micro litre trypan blue before loading it in hemocytometer.
Hi Nidhi,
Ok so average cells counted is 28 cells, you’re counting in the corner squares which means you have to multiply by 10,000. You’re doing a 1:1 dilution with trypan blue so the dilution factor is 2. The density of larvae in the original solution is 28 x 10,000 x 2 = 560,000 larvae / mL. The total amount in 80uL is then: 560,000 larvae/mL x 0.08 mL = 44,800 larvae. If you took 5uL from there to count (unless 80uL is after taking the 5uL), the actual amount left will be: 560,000 larvae/mL x 0.075 mL = 42,000 larvae.
Hope that was helpful!
Maria
Thank u so ma’am will contact you again in case of any problem.
Hi,
I’m counting sporangia (fungal fruiting bodies) in my suspension.
Their size is much bigger than a blood cell!
So, when I load onto a haemocytometer the usual way…. from the notch with the coverslip in place… I notice that majority of the sporangia…. are localized around the loading area and around the edges… only 1 or 2 sporangia… are seen in the grid area (nor an even distribution).
So I modified the loading techniques… by placing a drop of sporangia suspension (approx. 30ul) directly on the grid and then placing the coverslip… being careful to not create a spill over.
I could see an even distribution of sporangia on the slide area and on the grid…. And I could count well.
So, now the question is “ is the loading technique correct”….. the logic being the volume between the slide and coverslip should remain the same in both techniques??? Please let me know if your opinion!!!
Thanks
Jacob
Hi Jacob,
What you explain is definitely not the standard procedure to count cells, as it may result in a different volume being present under the coverslip and inaccurate cell concentrations calculated afterwards.
How big are the sporangia you are counting? I’ve read some of them may measure up to 60 μm, and they may produce aggregates. If that’s the case, it may be that the size makes it hard for them to move within the 100 μm space between the coverslip and chamber. Some of the suggestions here are to vortex them so they create a single-cell suspension, or using Tween agents to chemically separate them.
Cheers,
Maria
Hey Maria,
Thanks for such useful information.
I’ve tried to count yeast (S. cerevisiae) cells but everytime i load them in the chamber they are stick together due to budding (which is algo due favorable growth conditions) but it is hard to count them at this stage which i’ve noticed last for over 10h.
So do you have any recommendation for count them at this stage?
Thanks in advance
Juan
Hi Juan,
Giving your cells a good mix by vortexing should break them out of their bigger clumps. You may still see some clumps, in which case just try to estimate the number in the clump. And if you’re worried about that increasing your error, you can also count more squares. In my graduate work, I used the four large corner squares, rather than the central squares as discussed in the post. This would give you a greater sample area and thus reduce your error over using the central square alone.
This answer is late, but I hope it helps!
-Nick
Are there any established rules for counting fission yeasts? You mention different advice above, but I’ve heard rules for budding yeasts where you can count a bud as an individual yeast if it is 50% the size of the parent. I cannot seem to find any advice for fission yeast.
Fission yeasts seem to hang out for a while before they divide and when I use a conservative counting rule, I feel like I’m getting fermentation kinetics associated with a higher yeast count.
Thanks. -Stephen
That’s a great question. Based on https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5074380/ I might guess that “complete septum” would be a good rule? But if you’re unsure, you could count single and dividing cells separately and maybe that would capture the dynamics more fully for your needs? At the very least you’d be able to quantify the degree of error you might have depending on which rule you decide on.
HI. Just found your site now. I am a kombucha brewer and I am interested in finding the best way to count my kombucha culture (bacteria + yeats). What would you recommend?
Hope to hear from you soon. Cheers,
Hi Adriana!
A standard Improved Neubauer-style hemocytometer should be fine. You may want to use the center grid for the bacteria, with a higher microscope objective, so you can see them.
However, optical density, absorbance of light at 600 nm, is a fine way to measure culture growth in a coarser way. You might find that you can mainly use this, but spot check exact counts with a hemocytometer. I’m not sure what sort of equipment is available for that for brewers, but I believe some hobbyist DIY plans are available on the internet, to build your own.
Best,
Nick
hello, i want to ask, in my conditions, I ave cell pellet. one i directly dilute in 20uL culture medium and add 20uL of tryphan blue, while other I resuspend in 50uL pellet first and I take from them 10uL to mix with 10uL tryphan blue, so how can I calculate my total cells number ?
In both cases you have a dilution factor of 2 to get the original concentration prior to trypan blue. But if you want total cells instead of volume, in the latter case you need to multiply the concentration times the volume before adding the trypan blue.